
Energiestofwisselingsziekten
Elke week worden er kinderen geboren met een energiestofwisselingsziekte. Een genadeloze ziekte, waar nog geen medicijn voor is, maar het onderzoek daarnaar is in volle gang. Het onderzoek naar een medicijn is van levensbelang voor alle kinderen en volwassenen die deze ziekte hebben of in de toekomst deze vreselijke diagnose krijgen. Bij de ernstige vormen van de ziekte overlijdt een deel van deze kinderen al voor hun tiende levensjaar. Maar niemand kan bij de diagnose met zekerheid zeggen hoe het ziektebeeld zal verlopen en hoe oud het kind bijvoorbeeld kan worden.
De termen energiestofwisselingsziekten en mitochondriële ziekten worden gebruikt om een grote groep van ziekten te omschrijven waarvan de oorzaak ligt in de omzetting van voedingsstoffen naar energie.
Mitochondriën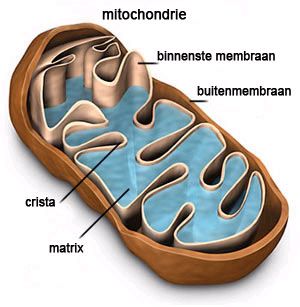
Bij een energiestofwisselingsziekte, ook wel Mitochondriële ziekte of Mito genoemd, gaat er wat mis in de energiestofwisseling van het lichaam. In elke cel zitten mitochondriën die we ook wel de energiefabrieken van de cel noemen. Mitochondriën kunnen energie uit suikers, vetten en eiwitten omzetten in een voor de cel bruikbare vorm van energie. Cellen die veel energie nodig hebben, zoals spiercellen, hebben daarom veel mitochondriën. Als deze energiefabrieken niet goed werken ontstaat er een energietekort en daardoor ontstaan er veel verschillende klachten met vaak ingrijpende gevolgen.
Klachten
Een energiestofwisselingsziekte is vaak een vrij onzichtbare ziekte. Patiënten zien er van buiten vaak goed uit, maar van binnen gaat er veel mis. Het aantal symptomen en de mate van ernst kan van persoon tot persoon verschillen. Dit hangt onder andere af van het soort fout in het erfelijk materiaal die de ziekte veroorzaakt en van het aantal zieke mitochondriën. De klachten variëren van ernstige vermoeidheid tot meervoudige handicaps, zowel lichamelijk als geestelijk. Ze kunnen meestal niet lang lopen, waardoor zij aangewezen zijn op een rolstoel. En door de spierzwakte zijn er problemen met slikken en krijgen ze sondevoeding om toch voldoende voeding binnen te krijgen. Als ook de hersenen en hartspier aangetast zijn, kan dit onder andere leiden tot een geestelijke handicap, epilepsie en/of hartproblemen. Dit is slechts een greep uit de verschillende klachten die zich voordoen bij deze ziekten. Duidelijk is wel dat het aantal klachten meestal toeneemt naarmate patiënten ouder worden. Door de variatie in klachten is het voor een arts vaak lastig een diagnose te vinden.
Diagnose
Mitochondriële ziekten kunnen op meerdere manieren worden onderverdeeld. De uiteindelijke diagnose wordt gesteld aan de hand van een combinatie van klinische klachten en verschijnselen, laboratorium onderzoeken (waaronder biochemisch onderzoek van de spier en huidcellen en DNA onderzoek). Onder het DNA onderzoek verstaan we zowel het DNA van de mitochondriën zelf (het mtDNA), als ook het kernDNA. Bij niet alle patiënten is op dit moment de onderliggende DNA fout gevonden. Nieuwe toegepaste technieken, zoals exoom en genoom analyse, zullen hier verandering in kunnen gaan brengen. Het bewijzen dat een DNA verandering ziekmakend is kan overigens vele jaren duren.
Energiestofwisselingsziekten kunnen als volgt worden ingedeeld:
a. Klinisch
Dit betreft de welomschreven ziektebeelden zoals de ziekte van Leigh, het MELAS syndroom, MIDD, MERRF, het Kearns-Sayre syndroom, Sengers syndroom, MEGDEL syndroom en Pearson syndroom.
b. Biochemisch
In een stukje spier of huid kan de omzetting van 'suiker' naar energie in haar geheel in een keer gemeten worden. Dit kan bijvoorbeeld met pyruvaat. We spreken van een pyruvaat-oxidatie stoornis als deze omzetting verstoord is. In datzelfde stukje spier of huid kan ook de vorming van hulpstoffen in de productie van energie (ATP) in de tijd gemeten worden. Dit noemen we de ATP productie snelheid. Tenslotte kunnen in het stukje spier of huid eiwitten, zoals enzymen, gemeten worden. De enzymen, waarvan er heel veel in de mitochondriën aanwezig zijn, zijn de hulpstoffen in de productie van energie (ATP). Op basis van bovenstaande kunnen patiënten biochemisch geclassificeerd worden als:
- patiënten met een gestoorde pyruvaat-oxidatie en ATP productie
- patiënten met een gestoorde pyruvaat-oxidate en ATP productie en met een of meer verlaagde activiteiten van enzymen of transporters betrokken bij de ATP productie. Voorbeelden hiervan zijn patiënten met een geïsoleerde complex I deficiëntie, of met een gecombineerde complex I en IV deficiëntie.
c. Moleculair
Onder een moleculaire classificatie verstaan we de indeling op DNA. We onderscheiden hierbij defecten in het mitochondriële DNA (mtDNA) of het kernDNA (nDNA). Sommige DNA fouten hebben ook een klinische naam gekregen zoals het Leigh syndroom. Inmiddels zijn er vele honderden fouten in beide DNAs beschreven die tot een mitochondriële ziekte aanleiding geven. Er zijn ook een groot aantal DNA veranderingen waarvan dit nog niet bewezen is. Veelal betreft dit stukjes DNA (gen) waarvan de functie niet of niet geheel bekend is. Soms betreft dit een bekend gen welke meer dan een functie blijkt te hebben. Soms betreft dit een gen geassocieerd met een bekende ziekte waarvan niet bekend was dat deze ook een verstoring van de energieproductie geeft.
Het stellen van de diagnose mitochondriële ziekte is niet eenvoudig. Expertcentra zoals het Radboud Centrum voor Mitochondriële Geneeskunde wegen zorgvuldig alle bovenstaande factoren om tot een definitieve diagnose te komen. Lees hier meer over op www.rcmm.info
Beloop van de ziekte
Er is een grote variatie in de leeftijd waarop mitochondriële ziekten zich kunnen presenteren (vrijwel direct naar de geboorte tot op latere volwassen leeftijd) als ook in ernst en het beloop van de ziekte. Sommige mitochondriële ziekten zijn zo ernstig dat kinderen jong overlijden. Bij anderen kan het beloop relatief milder zijn met ook qua levensverwachting een veel betere prognose. Niet altijd is op basis van klinisch, biochemisch of moleculair onderzoek is hier een goede inschatting van te maken en zal het beloop van de ziekte uiteindelijk meer duidelijkheid geven.
Nog geen medicijn
Er is nog geen enkel medicijn beschikbaar om energiestofwisselingsziekten te stoppen of te genezen. Omdat deze ziekten nog relatief onbekend en zeldzaam zijn, ziet de farmaceutische industrie letterlijk geen brood in het ontwikkelen van een medicijn. Voor professor Jan Smeitink en zijn team is dit onaanvaardbaar. Zij hebben er hun levenswerk van gemaakt om een medicijn te ontwikkelen om deze genadeloze ziekte te stoppen. Er zijn de laatste jaren grote stappen gezet en inmiddels is er een potentieel geneesmiddel dat getest wordt op de mens. Stichting Energy4All is met structurele financiële bijdragen erg belangrijk geweest in de versnelling van het onderzoek en dat zal zo blijven tot het medicijn er is voor patiënten met een energiestofwisselingsziekte.